Kiểm soát chất lượng dữ liệu (Cell Quality control)
1. Giới thiệu
Một khi đã có dữ liệu, chúng ta cần kiểm tra và loại bỏ các tế bào có chất lượng dữ liệu kém. Sai sót trong việc loại bỏ các tế bào có dữ liệu kém có thể làm nhiễu dữ liệu và ảnh hưởng tới việc tìm ra các thông tin mang ý nghĩa sinh học.
Hiện tại chưa có quy trình chuẩn cho thí nghiệm scRNA-seq, vậy nên các chỉ số kiểm tra chất lượng có thể biến đổi giữa các thí nghiệm. Khi kiểm tra chất lượng dữ liệu, chúng ta sẽ tìm những tế bào nằm ngoài vùng phân bố của phần lớn các tế bào còn lại. Lưu ý, khi so sánh chất lượng dữ liệu từ các quy trình khác nhau, chúng ta cần phải cân nhắc cẩn trọng.
2. Đọc dữ liệu
pbmc <- readRDS(file = "PBMC_1k.RDS")
3. Lọc bỏ tế bào kém chất lượng
Bước đầu trong kiểm tra chất lượng Seurat object là dựa vào sự biểu hiện tương đối của các gene từ ti thể (mitochodrial gene) để loại bỏ các tế bào không còn nguyên vẹn. Khi tế bào bước vào chu trình chết tự nhiên gây ra bởi “stress”, ti thể bên trong tế bào bị rò rỉ và RNA bị phá hỏng. Trong ví dụ này, chúng ta sẽ tính toán phần trăm các mitochrondial gene trên mỗi tế bào, thêm vào bảng dữ liệu, và biểu diễn chỉ số này bằng biểu đồ violin.
head(brain@meta.data)
orig.ident nCount_RNA nFeature_RNA
AAACCCAAGGAGAGTA-1 SeuratProject 8288 2620
AAACGCTTCAGCCCAG-1 SeuratProject 5512 1808
AAAGAACAGACGACTG-1 SeuratProject 4283 1562
AAAGAACCAATGGCAG-1 SeuratProject 2754 1225
AAAGAACGTCTGCAAT-1 SeuratProject 6592 1831
AAAGGATAGTAGACAT-1 SeuratProject 8845 2048
Ở đây, chúng ta tính toán phần trăm đoạn đọc của mitochondria gene và thêm 1 cột tên “percent.mito” vào meta.data của Seurat object. Như vậy, chúng ta có thể dùng phương trình violin plot để biểu diễn “percent.mito”.
Tìm mitochondiral genes bắt đầu bằng MT-
mt.genes <- rownames(pbmc)[grep("^MT-",rownames(pbmc))]
Lấy expression matrix của các mitochondrial gene này từ “counts” data trong Seurat objects
C<-GetAssayData(object = pbmc, slot = "counts")
Tính phần trăm mt genes và đưa số liệu này vào bảng metadata
percent.mito <- Matrix::colSums(C[mt.genes,])/Matrix::colSums(C)*100
pbmc <- AddMetaData(pbmc, percent.mito, col.name = "percent.mito")
Phương pháp tương tự được sử dụng để tính toán phần trăm ribosomal gene
rb.genes <- rownames(pbmc)[grep("^RP[SL]",rownames(pbmc))]
percent.ribo <- Matrix::colSums(C[rb.genes,])/Matrix::colSums(C)*100
pbmc <- AddMetaData(pbmc, percent.ribo, col.name = "percent.ribo")
Chúng ta có thể biểu diễn các giá trị này bằng biểu đồ violin. Ví dụ:
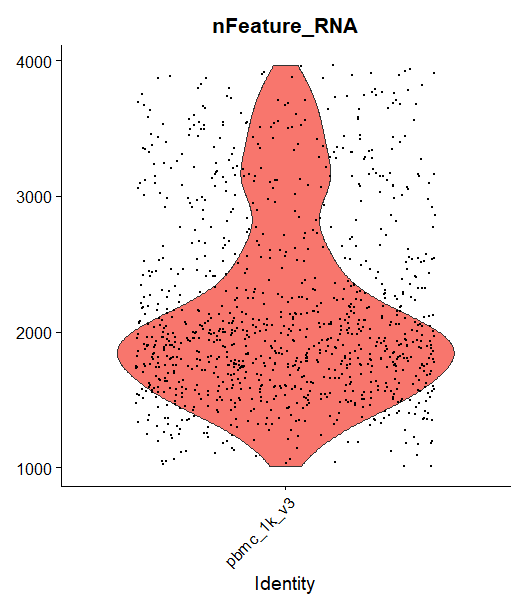
Biểu diễn số lượng gene (nFeature_RNA)
VlnPlot(pbmc, features = "nFeature_RNA", pt.size = 0.1) + NoLegend()
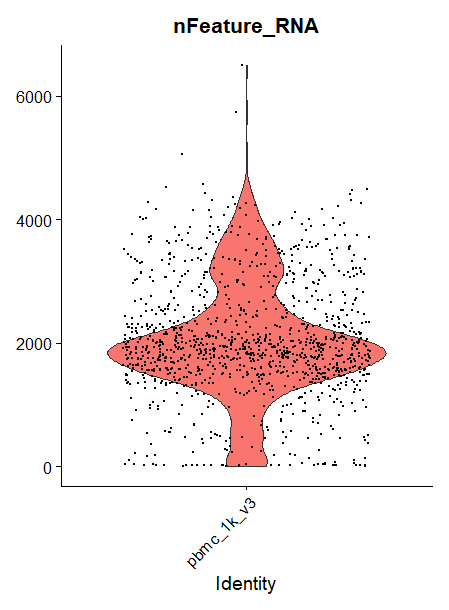
Biểu diễn số lượng UMIs (nCount_RNA)
VlnPlot(pbmc, features = "nCount_RNA", pt.size = 0.1) + NoLegend()
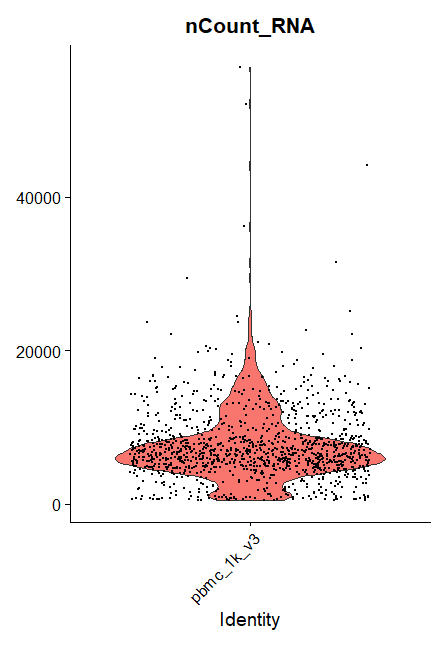
Biểu diễn phần trăm mitochondrial gene (percent.mito)
VlnPlot(pbmc, features = "percent.mito", pt.size = 0.1) + NoLegend()
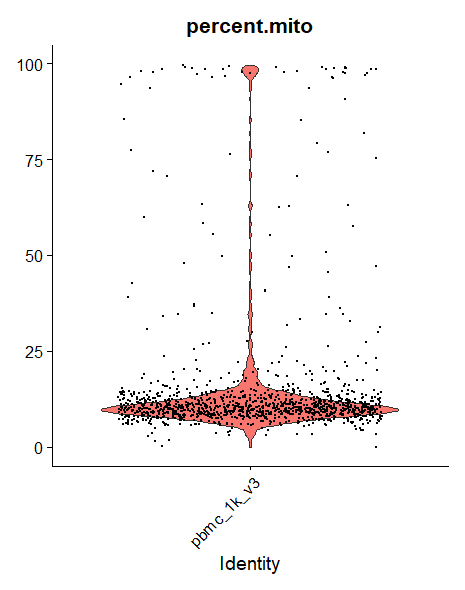
Biểu diễn phần trăm ribosomal gene (percent.ribo)
VlnPlot(pbmc, features = "percent.ribo", pt.size = 0.1) + NoLegend()
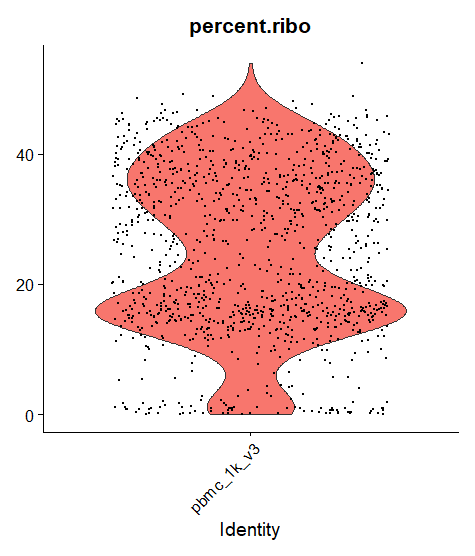
Kết hợp các biểu đồ thành một
FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")

FeatureScatter(pbmc, feature1 = "nFeature_RNA", feature2 = "percent.mito")

Sau khi kiểm tra chất lượng của tế bào, chúng ta sẽ loại bỏ các tế bào không đảm bảo tiêu chí. Ví dụ, chỉ lấy các tế bào có số lượng gene/tế bào lớn hơn 1000 và nhỏ hơn 4000, và các tế bào có phần trăm mitochondrial gene/tế bào nhỏ hơn 25%
pbmc <- subset(pbmc, subset = nFeature_RNA > 1000 & nFeature_RNA < 4000 & percent.mito < 25)
Kiểm tra lại data sau khi loại bỏ các tế bào kém chất lượng
VlnPlot(pbmc, features = "nFeature_RNA", pt.size = 0.1) + NoLegend()
Nguồn:
https://nbisweden.github.io/excelerate-scRNAseq/session-qc/Quality_control.html