Giảm chiều dữ liệu (Dimensionality reduction)
Phân tích thành phần chính (Principal component analysis - PCA)
Các phương pháp giảm kích thước tìm cách lấy một tập hợp lớn dữ liệu và trả về một tập hợp nhỏ hơn với các thành phần vẫn chứa hầu hết thông tin trong tập dữ liệu gốc.
Một trong những hình thức giảm kích thước đơn giản nhất là PCA. Phân tích thành phần chính (PCA) là một phương pháp toán học giúp biến đổi một số biến số tương quan (ví dụ: gene expression) thành một số (nhỏ hơn) các biến không tương quan được gọi là thành phần chính (“PC”).
Về mặt toán học, các PC tương ứng với các vector riêng (eigenvector) của ma trận hiệp phương sai. Các eigenvector được sắp xếp theo trị riêng (eigenvalue) để thành phần chính đầu tiên chiếm càng nhiều sự thay đổi (variability) trong dữ liệu càng tốt và mỗi thành phần tiếp theo lần lượt có phương sai cao nhất có thể với điều kiện là nó trực giao với các thành phần trước đó.
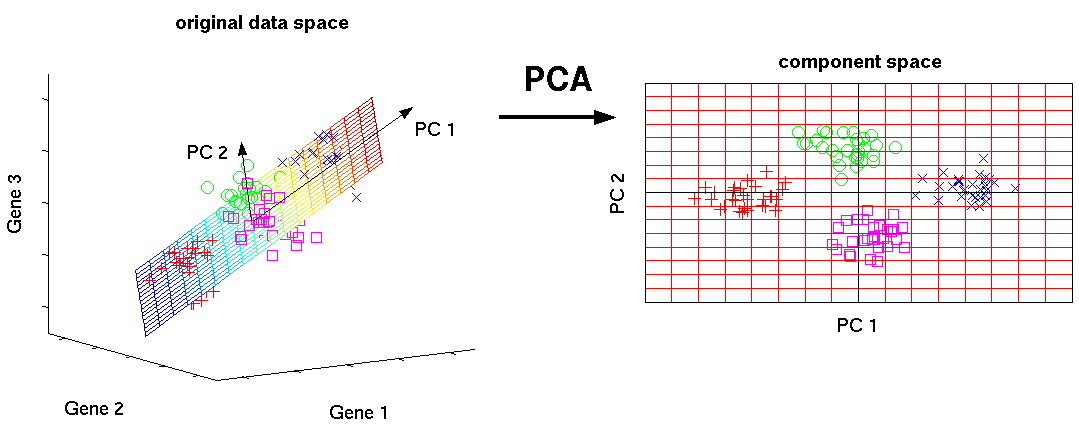
Về mặt sinh học, phương pháp giảm kích thước này hữu ích và thích hợp với dữ liệu scRNAseq vì tế bào phản ứng với môi trường của chúng bằng cách kích hoạt các chương trình điều hòa dẫn đến sự biểu hiện của mô-đun/gen. Kết quả là, sự biểu hiện gene hiển thị sự đồng biểu hiện có cấu trúc và giảm số chiều bằng cách phân tích các gene đồng biến đổi đó thành các thành phần chính, được sắp xếp theo mức độ phương sai mà chúng giải thích được.
Khuyến khích các bạn đọc thêm bài này: Link - Machine learning cơ bản
Thực hiện giảm số chiều tuyến tính
Tham khảo phiên bản Seurat v3: các feature (cách gọi thay thế cho gene khi đưa vào mô hình toán học) có tính biến số cao (highly variable - có nghĩa là các gene này có độ biến đổi cao, biểu hiện thấp ở một số tế bào này, nhưng biểu hiện cao ở các tế bào khác) được phát hiện thông qua hàm FindVariableFeatures đã được thực hiện ở phần tiền xử lý dữ liệu. Chúng ta sẽ sử dụng các feature này khi đã được scale để thực hiện PCA.
pbmc <- RunPCA(object = pbmc, npcs = 30, verbose = FALSE)
Seurat v3 cung cấp các chức năng để trực quan hóa PCA:
- Biểu đồ PCA
- Biểu đồ PCA được tô màu bởi một đối tượng định lượng (i.e gene)
- Biểu đồ phân tán (scatter)
- Biểu đồ Violin và Ridge
- Biểu đồ nhiệt
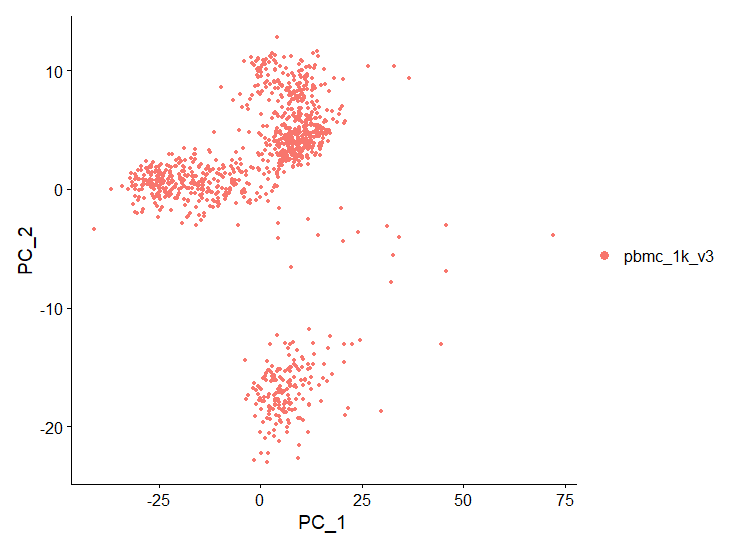
# Examine and visualize PCA results a few different ways
DimPlot(object = pbmc, reduction = "pca")
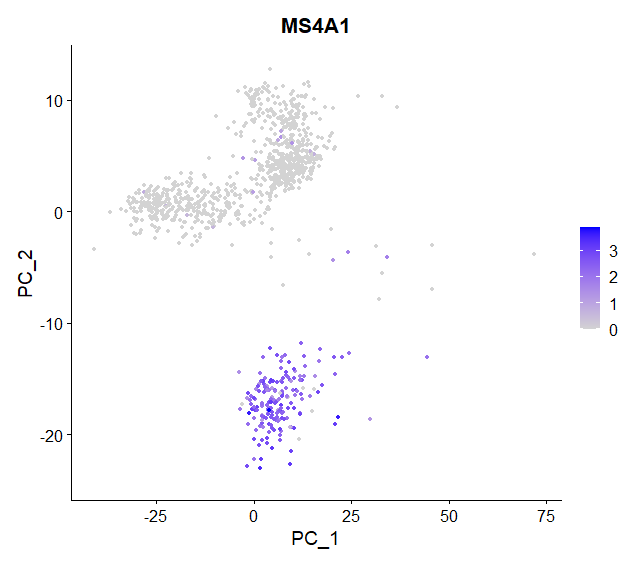
# Dimensional reduction plot, with cells colored by a quantitative feature
FeaturePlot(object = pbmc, features = "MS4A1")
Đặc biệt, DimHeatmap cho phép dễ dàng khám phá các nguyên nhân chính của sự không đồng nhất trong tập dữ liệu và nó rất hữu ích khi quyết định PC nào nên đưa vào cho các phân tích tiếp theo. Cả tế bào và gene đều được sắp xếp theo điểm số PCA của chúng. PCA là một phân tích rất có giá trị để khám phá các nhóm gene tương quan với nhau.
Xác định các thành phần chính có ý nghĩa thống kê
Để tránh trường hợp có quá nhiều nhiễu do lỗi kĩ thuật trong dữ liệu scRNAseq, Seurat phân cụm tế bào dựa trên PCA score, với mỗi PC đại diện cho một “metagene” kết hợp một tập hợp các gene có liên quan với nhau. Vì vậy, bước xác định số lượng PC dùng trong những phân tích tiếp theo là rất quan trọng.
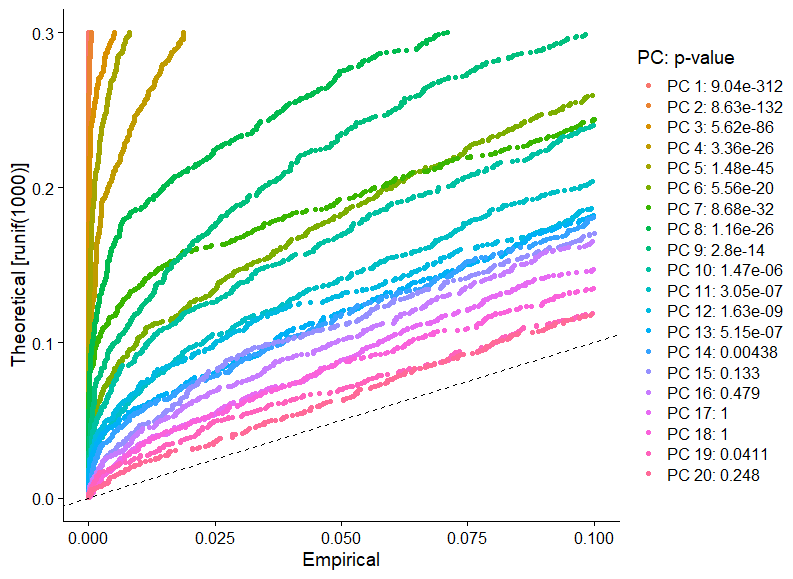
Trong Macoske et al., chúng ta tiến hành phép thử “resampling” (lấy mẫu lại), từ jackStraw procedure. Chúng ta tạo ngẫu nhiên một tập dữ liệu nhỏ (1% by default), chạy PCA, tạo một “null distribution” của biểu hiện gene, và lặp lại quá trình này. Từ phép thử này, chúng ta xác định số lượng PCs biểu diễn nhiều gene có giá trị p-value thấp.
# NOTE: This process can take a long time for big datasets, comment out for
# expediency. More approximate techniques such as those implemented in
# PCElbowPlot() can be used to reduce computation time
pbmc <- JackStraw(object = pbmc, reduction = "pca", dims = 20, num.replicate = 100, prop.freq = 0.1, verbose = FALSE)
pbmc <- ScoreJackStraw(object = pbmc, dims = 1:20, reduction = "pca")
JackStrawPlot(object = pbmc, dims = 1:20, reduction = "pca")
Phương pháp dùng để xác định số lượng PCs là vẽ biểu đồ biểu diễn “standard deviation” của PCs và xác định giá trị “cutoff” tại điểm gấp khúc trong đồ thị. Biểu đồ này có thể được thực hiện bởi hàm ElbowPlot. Trong ví dụ phía dưới, điểm gấp khác nằm xung quanh PC 5.
ElbowPlot(object = pbmc)

Chạy phương pháp giảm số chiều không tuyến tính bằng phương pháp tSNE
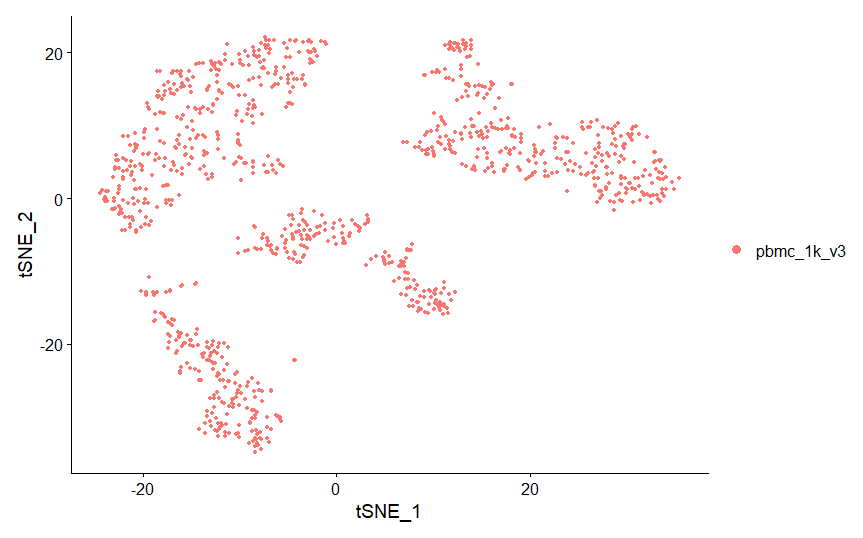
Một phương pháp khác để biểu diễn PCA là tSNE plot. tSNE (t-Distributed Stochastic Neighbor Embedding) kết hợp phương pháp giảm chiều dữ liệu (dimensionality reduction), ví dụ như PCA với random walk trong nearest-neighbour network kết nối dữ liệu nhiều chiều với một không gian 2 chiều. Khác với PCA, tSNE có thể thể hiện các cấu trúc không tuyến tính trong dữ liệu, và lưu giữ lại khoảng cách cục bộ giữa các tế bào. Do sự không tuyến tính và ngẫu nhiên (stochastic) của phương trình, tSNE thường khó để hiểu hết. Trong khi tSNE biểu diễn mối quan hệ cục bộ (local relationships) một cách rõ nét, thì nó lại không thể phản ánh mỗi quan hệ giữa các tế bào ở xa một cách chính xác. tSNE là một phương trình có tính ngẫu nhiên, có nghĩa là nếu chạy tSNE trên cùng một dữ liều nhiều lần, nó sẽ cho ra kết quả khác nhau. Để đảm báo tính lặp lại, trong dòng lệnh phía dưới, chúng ta sẽ “seed” một “random-number generator” để đảm bảo kết quả luôn cho ra một đồ thị.
pbmc <- RunTSNE(object = pbmc, dims.use = 1:10, do.fast = TRUE)
# note that you can set do.label=T to help label individual clusters
DimPlot(object = pbmc, reduction = "tsne")
Chạy phương pháp giảm số chiều không tuyến tính bằng phương pháp UMAP
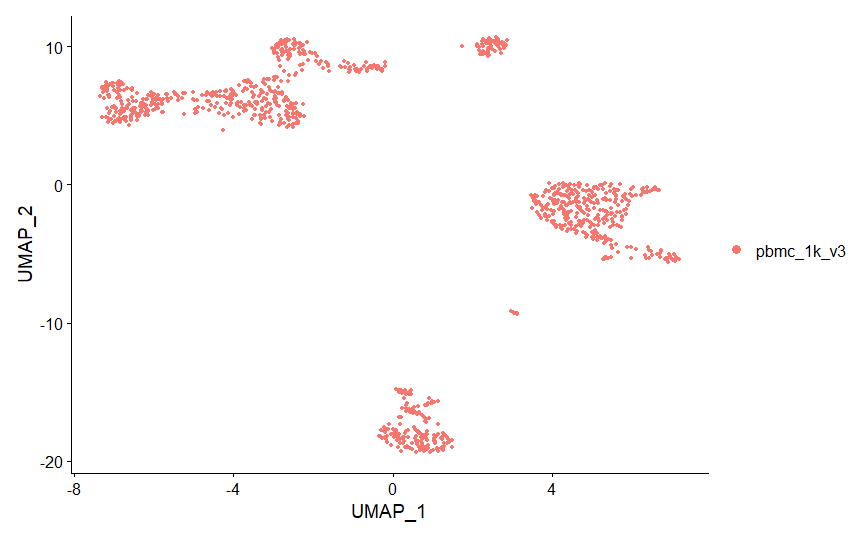
UMAP (Uniform Approximation and Projection) là một phương pháp giảm chiều không tuyến tính khác. Giống như tSNE, UMAP cũng bất biến và cần “random see generator” để đảm bảo tính lặp lại. Trong khi tSNE tối ưu hoá cho cấu trúc cục bộ, UMAP cần bằng giữa bảo toàn cấu trúc cục bộ và toàn bộ. Vì vậy, chúng ta sẽ sử dụng UMAP thay vì tSNE trong phân tích và biểu diễn dữ liệu.
pbmc <- RunUMAP(pbmc, reduction = "pca", dims = 1:20)
DimPlot(pbmc, reduction = "umap")
Nguồn:
https://chanzuckerberg.github.io/scRNA-python-workshop/analysis/03-dimensionality-reduction.html